








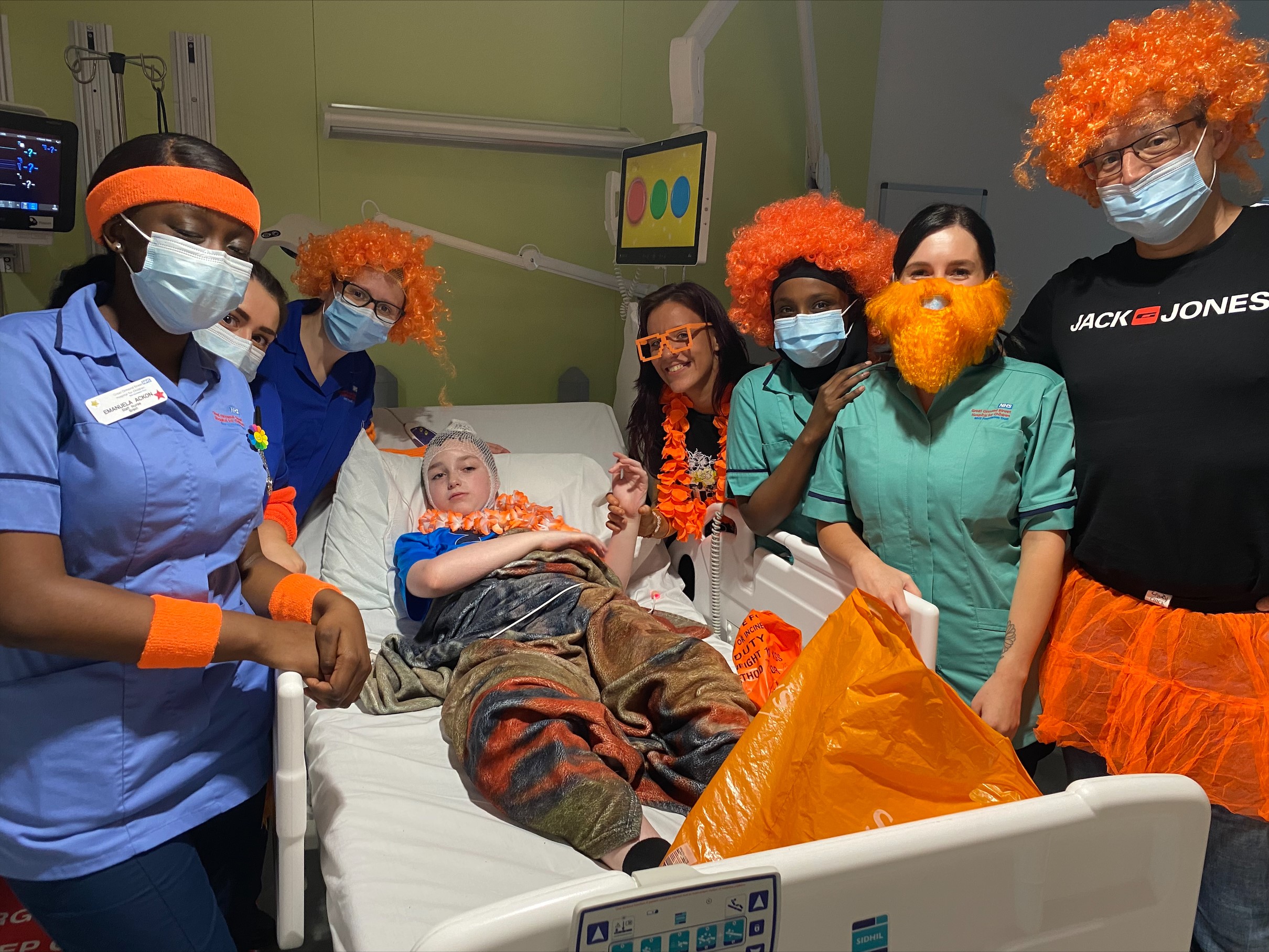




Our Mission
The BDFA’s mission is to enable everyone who is affected by Batten disease to live life to the full and secure the care and support they need until we find a cure.
The BDFA offers informed guidance and supports families and professionals who work with them as well as actively increasing awareness of the disease and funding future research to identify potential therapies and ultimately a range of cures.
The BDFA works across the rare disease sector to influence change in policy and processes as a collective voice.
Latest News
Help support Short Lives Cant Wait by signing the letter to the Prime Minister
Together for Short Lives has launched their ‘Short Lives can’t wait’ campaign this evening and we are encouraging families in the Batten community to get involved. Seriously, ill children and…

Joint Statement to the Global Batten Disease Community regarding update on Lexeo’s CLN2 disease gene therapy program
Dear Batten community, Over the past few years, Lexeo Therapeutics has been involved in developing an AAV-mediated gene therapy program for treating CLN2 Batten disease. However, additional funding and resources…

Invite your MP to Child Trust Fund Westminster Hall debate, important debate in Parliament next Tuesday 19 March at 4.30pm
Update from Disabled Children’s PartnershipThe amazing parent campaigner Andrew Turner has secured a Westminster Hall debate on Child Trust Funds for disabled young people. This is happening next Tuesday…

Next quarterly Town Hall Meeting
Dear Families,The BDFA Team and Trustees would like to invite you to our next quarterly Town Hall Meeting at 7pm on Wednesday 13th March.The purpose of the meeting is to…

Today, we stand together for Rare Disease Day! 29th February 2024
Let's raise awareness and support for those living with rare diseases. Every person battling a rare condition is a warrior, showing incredible strength, resilience, and determination in the face of…
Latest News

NEXT DAD’S CHAT, Wednesday 28th February at 8pm
A chance to come together on Zoom with other dads from the BDFA community and have a chat.Led by trained Peer Befriender Andrew Dawkins and James Yarrow. If you are…

Theranexus Update for CLN3 Programme
Dear Families, You may be aware that there was a press release last week from Theranexus about the Batten-1 clinical trial for CLN3 (Theranexus_PR_Cash_Position_Dec_31_2023_VDEF.pdf). Beyond Batten have followed this up…
Taysha Gene Therapies Provides Update on Deprioritized Pipeline Programs
Joint Statement to the Global Batten Disease Community On Thursday, February 15, 2024, important news broke regarding CLN1 clinical research that global Batten disease patient advocacy groups wish to share…

International Epilepsy Day
It’s International Epilepsy Day - a day for coming together to raise awareness. Batten Disease is one of many rare and complex epilepsies affecting families in the UK. While everyone’s…

Joint Statement to the Global Batten Disease Community regarding the future of the CLN3 and CLN6 gene therapy clinical programs led by Amicus Therapeutics
Dear Batten community, As many of you know, we have been awaiting further news regarding the future of the CLN3 and CLN6 gene therapy clinical programs led by Amicus Therapeutics.…

Visit the online shop to help support the work of the BDFA






