
Progress in preclinical study of enzyme replacement therapy for the CLN1 form of Batten Disease
Professor Sandra Hofmann, M.D., Ph.D. UT Southwestern Medical Center, Dallas, USA
Biography
Dr. Hofmann’s laboratory has done pioneering work on the metabolism of proteins that are modified by lipids. This work has led to a greater understanding of a group of hereditary brain disorders, the neuronal ceroid lipofuscinoses (NCL, or Batten Disease). Her laboratory demonstrated that the most severe form of NCL is caused by a failure to metabolize fatty acid-modified proteins.
The CLN1 form of Batten disease is caused by build-up of material in the brain due to lack of an enzyme called palmitoyl-protein thioesterase, or PPT.
PPT – What is its function in the Lysosome?
The normal function of PPT is to break a chemical bond between naturally-occurring linkages between fatty acids and amino acids in proteins (the amino acid cysteine, in particular). The enzyme normally functions in a compartment within each cell (the lysosome) where degradation and recycling of cellular material normally occurs. Lack of the enzyme PPT is harmful to neurons and causes the serious manifestations of infantile Batten disease. Some older children with Batten Disease (CLN1-late infantile and CLN1-juvenile) also have PPT deficiency with a later onset due to mutations that provide a small amount (2-4%) of normal enzyme function. If PPT could be supplied back to the brain, it is possible that the loss of neurons could be stopped, but probably not reversed.
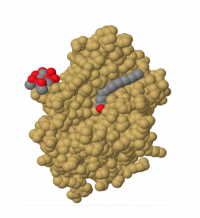
Atomic model of the PPT enzyme. The structure is derived from X-ray analysis of pure crystalline PPT protein. The gray atoms in the center are the fatty acid that PPT has just removed from a protein. The gray and red “knob” on the left side is the mannose 6-phosphate sugar that is recognized by cells and allows PPT to be taken into the cell and the lysosome.
Studies involving model systems for CLN1 Batten disease.
A mouse model was created that is genetically engineered to lack the PPT enzyme in order to allow scientists to study the disease. The mice become ill around 5 months of age and survive for 7 to 8 months. The normal lifespan of a mouse is about 2-3 years. The mouse model has been useful for testing therapies for Batten disease caused by lack of PPT.
Mice treated with PPT injected into the cerebrospinal fluid perform better in a test of motor function. Mice are placed on a rotating rod and the time it takes for the mouse to slip and fall off is recorded. Each test is ended at 60 seconds. Normal mice easily complete the test. Mice lacking PPT can only stay on the rod for about 5 seconds when they are 7 months old. Mice treated with PPT perform better on the test, and the higher the concentration of PPT in the injection, the better the performance.
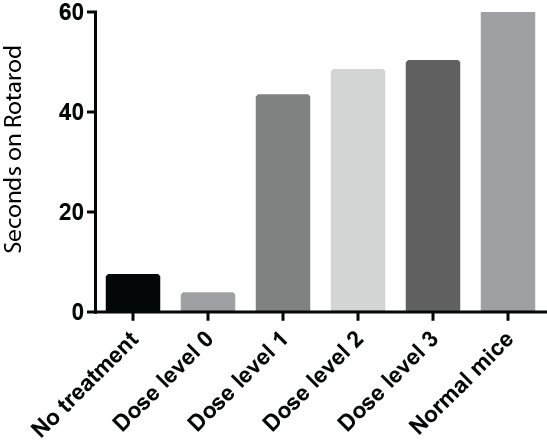
The BDFA funded a very important part of the preclinical work to determine the effect of administering PPT directly to the central nervous system of PPT deficient mice by injection into the cerebrospinal fluid, as in a spinal tap. About 80 mice were used for the experiment. Mice received no injections, injection of a solution of artificial cerebrospinal fluid, or the same fluid with three different dose levels of PPT. For technical reasons only one set of injections was performed, at around 6 weeks of age, which is early adulthood for a mouse. Injected mice were subjected to motor tests throughout the lifespan. Data gathered so far indicate that injected mice live longer, perform better on motor tests and have improvements in brain and spinal cord pathology.

Caption for. Drs. Jui-Yun (Laura) Lu and Sandra Hofmann, University of Texas Southwestern Medical Center received BDFA support for their work on CLN1/PPT1. Not shown is Dr. Lingling Wang, who performed all of the mouse injections for the study described in the article.
This work requires skill and injections must be completed according to an exact schedule on a daily basis. In the middle of all these 200 or so mouse injections required for the study, there was a major freak snowstorm in Dallas and it took Laura and Lingling 4 hours to get to the laboratory to continue the experiment; normally a 15 minute trip and quite an ordeal. They also ended up staying on the floor of a friend’s apartment across from the university!

Further Studies
Currently there is a clinical trial for children with CLN2 Batten Disease in which the deficient enzyme TPP-1 (BMN-190) is being administered to the cerebrospinal fluid of the brain. The clinical trial was based largely upon preclinical mouse studies involving the CLN2 enzyme, much like the current study of CLN1. Results of the current study funded by the BDFA on CLN1/PPT support using the same approach for CLN1 disease, particularly if BMN-190 shows good results for CLN2, late infantile NCL.
There is still much work to be done and future experiments would need to prove the safety of the PPT1 enzyme in at least two species, which is a requirement for regulatory agencies, and more detailed efficacy studies for the final therapeutic product will also need to be completed. Because the final therapeutic dose given to patients will need to be tested in a variety of ways to ensure safety and effectiveness, these studies are best done in the context of a pharmaceutical company drug development program. The results of the study funded by the BDFA will make infantile Batten disease/CLN1 an attractive opportunity for drug development.
Professor Hofmann and her team are going to publish the results of this study shortly and with this the BDFA will be working to ensure this important work is taken forward.